Kaletra
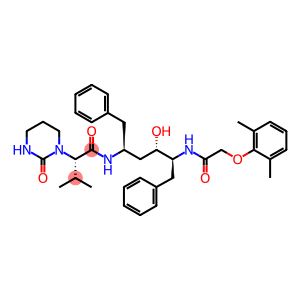
Lopinavir
CAS: 192725-17-0
Molecular Formula: C37H48N4O5
Kaletra - Names and Identifiers
Kaletra - Physico-chemical Properties
| Molecular Formula | C37H48N4O5 |
| Molar Mass | 628.81 |
| Density | 1.163±0.06 g/cm3(Predicted) |
| Melting Point | 255.2-260.6 °F (124—127°C) |
| Boling Point | 924.1±65.0 °C(Predicted) |
| Flash Point | 512.7°C |
| Solubility | DMSO: soluble20mg/mL, clear |
| Vapor Presure | 0mmHg at 25°C |
| Appearance | powder |
| Color | white to beige |
| pKa | 13.89±0.46(Predicted) |
| Storage Condition | 2-8°C |
| Stability | Hygroscopic |
| Refractive Index | 1.577 |
| Use | Lopinavir is a potent HIV protease(HIV protease) inhibitor with Ki of 1.3 pM. |
| In vitro study | Lopinavir binds to mutant HIV proteases (V82A, V82F, and V82T) with K I of 4.9 pM, 3.7 pM, and 3.6 pM, respectively. 0.5 nM Lopinavir inhibited 93% of wild-type HIV protease activity. Lopinavir acts on MT4 cells. In the presence or absence of 50% HS, Lopinavir inhibits HIV protease activity with EC50 of 102 nM and 17 nM, respectively. Lopinavir is converted into some metabolites in liver microparticles, the primary metabolites are M-3 and M-4, and this effect is NADPH-dependent. Lopinavir is a potent Rh123 inhibitor, acting on the Caco-2 cell layer with an IC50 of 1.7 mM. Lopinavir treatment of LS 180v cells for 72 hours, intracellular Rh123 content decreased. Lopinavir acts on LS 180v cells to induce P-glycoprotein immunoreactive protein and messenger RNA levels. Lopinavir inhibits the C subtype clone C6 with an IC50 of 9.4 nM. Lopinavir acts on human liver microsomes, inhibits CYP3A,IC50 is 7.3 mM, and weakly inhibits human CYP1A2,2B6,2C9,2C19,2D6. |
| In vivo study | Lopinavir was administered orally to rats at a dose of 10 mg/kg with a Cmax of 0.8 μg/mL and an oral bioavailability of 25%. |
Kaletra - Risk and Safety
| UN IDs | 3077 |
| HS Code | 29335990 |
Kaletra - Nature
Open Data Verified Data
crystalline solid. The melting point was 124-127 °c.
Last Update:2024-01-02 23:10:35
Kaletra - Preparation Method
Open Data Verified Data
oxalyl chloride was added dropwise to a solution of dimethyl sulfoxide and dichloromethane at a certain temperature. After the reaction, a dichloromethane solution of N-benzyloxycarbonyl monoaminopropanol was added, and after a reaction period of time, triethylamine was added dropwise and stirred. This was taken up in a cold 10% aqueous citric acid solution and extracted with ether. The organic layers were combined, washed with saturated brine, dried, filtered, and concentrated under reduced pressure. Aminopropionaldehyde derivatives were purified by Silica Gel chromatography. It was dissolved in methanol, valine methanol hydrochloride, sodium acetate and sodium cyanoborohydride were added, stirred at room temperature, concentrated under reduced pressure, dissolved in ethyl acetate, washed with saturated sodium bicarbonate, and extracted with ethyl acetate. The organic layers were combined, washed with saturated brine, dried, filtered and concentrated under reduced pressure. The resulting material was purified by Silica Gel chromatography to give a condensation reduction product. The product was subjected to hydrogenation and then reacted with 1,1-carbonyl bis (1-tetrahydro-pyrimidine) of ISO-Kul in dichloromethane. The resulting product was hydrolyzed with lithium hydroxide in water and dioxane, acidified with hydrochloric acid and extracted with ethyl acetate. The extract was dried, filtered and concentrated under reduced pressure to give 2 S-(tetrahydropyrimidin-2-ox-1-yl) 3-methylbutyric acid. Dissolve it and (2S,3S,5S) -2-(2.6-dimethylphenoxyacetyl) amino -3-hydroxy -5-amino -1,6-= phenylhexane in dimethylformamide, (3-dimethylaminopropyl)-3-ethylcarbodiimide was added and ropifenavir was reacted.
Last Update:2022-01-01 09:08:22
Kaletra - Preparation solution concentration reference
| 1mg | 5mg | 10mg | |
|---|---|---|---|
| 1 mM | 1.59 ml | 7.952 ml | 15.903 ml |
| 5 mM | 0.318 ml | 1.59 ml | 3.181 ml |
| 10 mM | 0.159 ml | 0.795 ml | 1.59 ml |
| 5 mM | 0.032 ml | 0.159 ml | 0.318 ml |
Last Update:2024-01-02 23:10:35
Kaletra - Use
Open Data Verified Data
developed by Abbott Laboratories and marketed in the United States on September 15, 2000. AIDS Virus protease inhibitors, antiretroviral drugs Virus. For the treatment of HIV-1 infections in adults and pediatric patients over 6 months.
Last Update:2022-01-01 09:08:23
Supplier List
Product Name: Lopinavir Request for quotation
CAS: 192725-17-0
Tel: +86 13313090628
Email: 13313091926@163.com
Mobile: +86 13313090628
Wechat: 13373390591
WhatsApp: 18733928930
CAS: 192725-17-0
Tel: +86 13313090628
Email: 13313091926@163.com
Mobile: +86 13313090628
Wechat: 13373390591
WhatsApp: 18733928930
Spot supply
Product Name: Lopinavir Visit Supplier Webpage Request for quotationCAS: 192725-17-0
Tel:
Email: qianyanbiochem@gmail.com
Mobile: 13247110337
QQ: 2972965813
Product List: View Catalog
Spot supply
Product Name: Lopinavir Visit Supplier Webpage Request for quotationCAS: 192725-17-0
Tel: +86-400-900-4166
Email: product@acmec-e.com
Mobile: +86-18621343501
QQ: 2881950922
Wechat: 19602116810
WhatsApp: +86-18621343501
Product Name: Lopinavir Request for quotation
CAS: 192725-17-0
Tel: +86 19943533199
Email: vikki@api-made.com
QQ: 535948114
WhatsApp: +86 19943533199
CAS: 192725-17-0
Tel: +86 19943533199
Email: vikki@api-made.com
QQ: 535948114
WhatsApp: +86 19943533199
Spot supply
Product Name: Lopinavir Visit Supplier Webpage Request for quotationCAS: 192725-17-0
Tel: 18301782025
Email: 3008007409@qq.com
Mobile: 18021002903
QQ: 3008007409
Wechat: 18301782025
Product Name: Lopinavir Request for quotation
CAS: 192725-17-0
Tel: +86 13313090628
Email: 13313091926@163.com
Mobile: +86 13313090628
Wechat: 13373390591
WhatsApp: 18733928930
CAS: 192725-17-0
Tel: +86 13313090628
Email: 13313091926@163.com
Mobile: +86 13313090628
Wechat: 13373390591
WhatsApp: 18733928930
Spot supply
Product Name: Lopinavir Visit Supplier Webpage Request for quotationCAS: 192725-17-0
Tel:
Email: qianyanbiochem@gmail.com
Mobile: 13247110337
QQ: 2972965813
Product List: View Catalog
Spot supply
Product Name: Lopinavir Visit Supplier Webpage Request for quotationCAS: 192725-17-0
Tel: +86-400-900-4166
Email: product@acmec-e.com
Mobile: +86-18621343501
QQ: 2881950922
Wechat: 19602116810
WhatsApp: +86-18621343501
Product Name: Lopinavir Request for quotation
CAS: 192725-17-0
Tel: +86 19943533199
Email: vikki@api-made.com
QQ: 535948114
WhatsApp: +86 19943533199
CAS: 192725-17-0
Tel: +86 19943533199
Email: vikki@api-made.com
QQ: 535948114
WhatsApp: +86 19943533199
Spot supply
Product Name: Lopinavir Visit Supplier Webpage Request for quotationCAS: 192725-17-0
Tel: 18301782025
Email: 3008007409@qq.com
Mobile: 18021002903
QQ: 3008007409
Wechat: 18301782025
View History