利奈唑胺
雷奈佐利(linezolid)
CAS: 165800-03-3
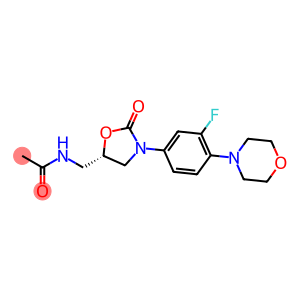
化学式: C16H20FN3O4
性质
白色结晶,熔点181.5~182.5℃。
制法
将乙酸乙酯、吗啉和二异丙基乙基胺混合,搅拌下滴加3,4一二氟硝基苯,反应完毕后。用乙酸乙酯萃取,饱和盐水洗涤,干燥,蒸干,丙酮一水重结晶,得3一氟-4-吗啉基硝基苯。将还原铁粉、水和冰乙酸混合,回流一定时间,滴加3一氟一4一吗啉基硝基苯的无水乙醇溶液,反应完毕后,趁热过滤,蒸干,得到的物质用乙酸乙酯萃取,盐水洗涤,干燥,蒸干,得3一氟-4-吗啉基苯胺。将其溶于丙酮和水的混合溶液中,加入碳酸氢钠,滴加氯甲酸苄酯,搅拌,抽滤,得固体,滤液放入冰水,抽滤得到固体。所得固体用丙酮一水重结晶,得苄氧羰基-3-氟-4-吗啉基苯胺。
将其溶于无水四氢呋喃,在氨气保护下,滴加丁基锂溶液,再滴加(R)-缩甘油丁酯,反应后,加入饱和氯化铵溶液、乙酸乙酯、水。用乙酸乙酯萃取,饱和盐水洗,干燥,蒸干,将得到的物质溶于二氯甲烷和三乙胺中,滴加甲磺酰氯进行反应。用二氯甲烷萃取,用饱和盐水洗,干燥,蒸干,得磺酸酯。将磺酸酯和叠氮化钠溶于二甲基甲酰胺反应。用乙酸乙醇萃取,饱和盐水洗,干燥,蒸干,取代得叠氮衍生物。将该衍生物和5%钯一碳在无水乙醇中反应一定时间,过滤,蒸干,得到的物质溶于四氢呋喃和氢氧化钠水溶液中,加入乙酸酐,用乙酸乙酯萃取,饱和盐水洗,干燥。蒸干,硅胶柱层析,乙酸乙酯一正己烷重结晶,即得利奈唑胺。
用途
美国法玛西亚( Pharmacia&Upj ohn)制药公司开发,2000年4月在美国首次上市。抗生素,为细菌蛋白质合成抑制剂。利奈唑胺对甲氧西林敏感或耐药葡萄球菌、万古霉素敏感或耐药肠
菌、青霉素敏感或耐药肺炎链球菌均显示了良好的抗菌作用,对厌氧菌亦具抗菌活性。与其他药物不同,利奈唑胺不影响肽基转移酶活性。利奈唑胺的作用部位和方式独特,因此在具有本质性或获得性耐药特征的阳性细菌中,都不易与其他抑制蛋白合成的抗菌药发生交叉耐药,在体外也不易诱导细菌耐药性的产生。用于耐万古霉素粪肠球菌感染、并发性和非并发性皮肤与皮肤结构感染。

利奈唑胺 - 性质
可信数据
白色结晶,熔点181.5~182.5℃。
最后更新:2024-01-02 23:10:35
利奈唑胺 - 制法
可信数据
将乙酸乙酯、吗啉和二异丙基乙基胺混合,搅拌下滴加3,4一二氟硝基苯,反应完毕后。用乙酸乙酯萃取,饱和盐水洗涤,干燥,蒸干,丙酮一水重结晶,得3一氟-4-吗啉基硝基苯。将还原铁粉、水和冰乙酸混合,回流一定时间,滴加3一氟一4一吗啉基硝基苯的无水乙醇溶液,反应完毕后,趁热过滤,蒸干,得到的物质用乙酸乙酯萃取,盐水洗涤,干燥,蒸干,得3一氟-4-吗啉基苯胺。将其溶于丙酮和水的混合溶液中,加入碳酸氢钠,滴加氯甲酸苄酯,搅拌,抽滤,得固体,滤液放入冰水,抽滤得到固体。所得固体用丙酮一水重结晶,得苄氧羰基-3-氟-4-吗啉基苯胺。
将其溶于无水四氢呋喃,在氨气保护下,滴加丁基锂溶液,再滴加(R)-缩甘油丁酯,反应后,加入饱和氯化铵溶液、乙酸乙酯、水。用乙酸乙酯萃取,饱和盐水洗,干燥,蒸干,将得到的物质溶于二氯甲烷和三乙胺中,滴加甲磺酰氯进行反应。用二氯甲烷萃取,用饱和盐水洗,干燥,蒸干,得磺酸酯。将磺酸酯和叠氮化钠溶于二甲基甲酰胺反应。用乙酸乙醇萃取,饱和盐水洗,干燥,蒸干,取代得叠氮衍生物。将该衍生物和5%钯一碳在无水乙醇中反应一定时间,过滤,蒸干,得到的物质溶于四氢呋喃和氢氧化钠水溶液中,加入乙酸酐,用乙酸乙酯萃取,饱和盐水洗,干燥。蒸干,硅胶柱层析,乙酸乙酯一正己烷重结晶,即得利奈唑胺。
最后更新:2022-01-01 08:52:49
利奈唑胺 - 介绍
Linezolid 是一种合成的抗生素,用于治疗严重感染。Prototype of the oxazolidinone antimicrobials and inhibits bacterial mRNA translation.
最后更新:2022-10-16 17:14:15
利奈唑胺 - 用途
可信数据
美国法玛西亚( Pharmacia&Upj ohn)制药公司开发,2000年4月在美国首次上市。抗生素,为细菌蛋白质合成抑制剂。利奈唑胺对甲氧西林敏感或耐药葡萄球菌、万古霉素敏感或耐药肠
菌、青霉素敏感或耐药肺炎链球菌均显示了良好的抗菌作用,对厌氧菌亦具抗菌活性。与其他药物不同,利奈唑胺不影响肽基转移酶活性。利奈唑胺的作用部位和方式独特,因此在具有本质性或获得性耐药特征的阳性细菌中,都不易与其他抑制蛋白合成的抗菌药发生交叉耐药,在体外也不易诱导细菌耐药性的产生。用于耐万古霉素粪肠球菌感染、并发性和非并发性皮肤与皮肤结构感染。
最后更新:2022-01-01 08:52:49
利奈唑胺 - 简介
雷奈佐利是一种有机化合物,化学名称为苯甲醛。它是一种无色液体,具有强烈的芳香味道。
雷奈佐利常用于香水和香料制造中,因为它具有持久的芳香特性。它也用作有机合成中的中间体,可用于合成各种化合物。
雷奈佐利的制法可以通过苯甲醇与氧气在催化剂存在下进行氧化反应得到。反应条件可以通过控制温度、氧气压力和反应时间来改变。
安全信息:雷奈佐利是一种易燃液体,应远离明火和高温。在使用和储存时,应采取防火措施,并且要避免与强氧化剂接触。接触皮肤或眼睛可能引起刺激,在操作时应佩戴适当的防护设备,如手套和护目镜。如果意外接触,应立即用大量清水冲洗并就医。在使用之前,请仔细阅读和遵守相关的安全数据表。
雷奈佐利常用于香水和香料制造中,因为它具有持久的芳香特性。它也用作有机合成中的中间体,可用于合成各种化合物。
雷奈佐利的制法可以通过苯甲醇与氧气在催化剂存在下进行氧化反应得到。反应条件可以通过控制温度、氧气压力和反应时间来改变。
安全信息:雷奈佐利是一种易燃液体,应远离明火和高温。在使用和储存时,应采取防火措施,并且要避免与强氧化剂接触。接触皮肤或眼睛可能引起刺激,在操作时应佩戴适当的防护设备,如手套和护目镜。如果意外接触,应立即用大量清水冲洗并就医。在使用之前,请仔细阅读和遵守相关的安全数据表。
最后更新:2024-04-09 20:52:54
供应商列表
利奈唑胺的上游原料
您刚刚浏览过
你知道吗?
点击右上角的地图标志可以切换至英文页面